
Sindrome di Turner
Che cos’è la sindrome di Turner
La Sindrome di Turner è la più frequente disgenesia gonadica a espressione fenotipica femminile. La Sindrome di Turner nella forma classica, è caratterizzata da amenorrea primaria, dalla presenza di gonadi rudimentali, infantilismo sessuale alla pubertà, bassa statura e anomalie congenite multiple. L’anomalia è riscontrata in circa il 20% degli aborti spontanei che si verificano nel primo trimestre di gravidanza. La mancanza del cromosoma X può essere dovuta sia alla perdita del cromosoma durante la gametogenesi, sia a un errore mitotico durante una delle suddivisioni precoci che avvengono nello zigote fecondato. Circa la metà delle pazienti possiede cariotipo 45,X, mentre circa un quarto di esse mostra un mosaicismo senza anomalie cromosomiche strutturali (46,XX/45,X). Infine un quarto dei soggetti affetti da sindrome di Turner possiede un cromosoma X strutturalmente anormale
(isocromosoma X,X ad anello, delezione parziale del braccio lungo o del braccio corto di X) con o senza mosaicismo. La sindrome di Turner è dovuta nei due terzi dei casi a perdita del cromosoma X paterno e solo nel 20% dei casi di quello materno. Non esiste alcuna correlazione tra età materna e comparsa della malattia. La bassa statura e le altre anomalie somatiche caratteristiche della sindrome di Turner sono conseguenti alla perdita di materiale genetico del braccio corto del cromosoma X, mentre i loci cromosomici la cui monosomia è responsabile della mancata maturazione gonadica sono tre, due sul braccio lungo e uno sul braccio corto del cromosoma X. La presenza di un doppio dosaggio allelico in questi loci appare essere indispensabile per un normale sviluppo della gonade femminile. In presenza del cariotipo 45,X durante l’embriogenesi l’abbozzo gonadico si forma con un numero di ovociti non particolarmente ridotto, ma l’atresia dei follicoli primordiali e primari è così accelerata e massiva che il loro numero è quasi totalmente azzerato alla nascita.
Quadro clinico
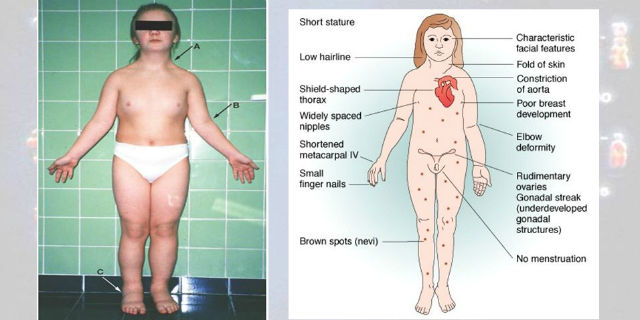
Le caratteristiche cliniche con le relative frequenze dei soggetti affetti da Sindrome di Turner.
Le stigmate somatiche sono rappresentate da:
- statura
- collo corto e palmato (pterigium colli)
- micrognazia
- epicanto
- torace a scudo
- valgismo del gomito
- brevità abnorme del quarto metacarpo
- unghie ipoplasiche
- palato ogivale
- orecchie sporgenti e impiantate in basso
- attaccatura bassa dei capelli.
L’insieme di queste malformazioni conferisce al volto delle pazienti con sindrome di Turner un caratteristico aspetto il cosiddetto volto a sfinge.
In alcuni soggetti si riscontrano anomalie congenite a carico del cuore e dei vasi, in particolare:
- stenosi aortica
- coartazione aortica
- anomalie a carico del rene
- rene a ferro di cavallo
A livello cutaneo:
- nevi pigmentati
- tendenza alla formazione di cheloidi
La maturazione scheletrica è normale o modestamente ritardata durante l’infanzia, ma è deficitaria durante l’adolescenza come conseguenza del deficit gonadico di steroidi. Spesso lo scheletro presenta aree di rarefazione, soprattutto alle mani, i piedi, ai gomiti e alla porzione superiore del femore. I pazienti non trattati con terapia estrogenica sostitutiva possonono sviluppare una severa osteoporosi. Frequenti sono anche lesioni similosteocondrosiche della colonna vertebrale e la scoliosi. Nei soggetti con sindrome di Turner sono descritte numerose associazioni con difetti di origine autoimmune quali:
- diabete mellito
- tiroide linfocitaria cronica
- artrite reumatoide
- obesità
- ipertensione arteriosa
- artrite reumatoide
- l’otite media recidivante
Nelle prime fasi di vita è frequente il riscontro di linfedema al collo, alle mani e ai piedi che, a volte, può costituire l’unico elemento per cui sospettare una sindrome di Turner. Tale linfedema regredisce spontaneamente nel tempo. Alla nascita queste bambine mostrano un peso inferiore alla media e un deficit della crescita staturale. La velocità di crescita può essere normale nelle prime fasi della vita, per poi ridursi drasticamente dai 8 anni in poi, e l’altezza finale risulta in media attorno ai 142 cm (range 132-152).
Le tube di Falloppio sono normalmente presenti, ma iposviluppati in conseguenza della ridotta produzione estrogenica da parte del rudimenti gonadici. Le gonadotropine circolanti, soprattutto l’ormone follicolostimolante (FSH), sono elevate. I valori di FSH presentano tipico andamento bifasico. L’FSH è, infatti, molto elevato nei primi 2-4 anni di vita, si riduce quindi dai 6 ai 10 anni (insieme valori di LH) raggiungendo livelli vicini alla normalità, per poi aumentare di nuovo. I valori di a AMH sono bassi/insondabili indicandola ridotta/assente riserva follicolare ovarica.
In circa il 70% dei casi soprattutto quelli con cariotipo 45,X o associati ad anomalie del secondo X, è presente amenorrea primaria. Negli altri casi è invece possibile osservare una pubertà spontanea seguita a volte da cicli regolari con ovulazione anche per alcuni anni. L’amenorrea secondaria con ulteriore deviazione FSH e azzeramento dell’ AMH circolante viene raggiunta entro i 25 anni di vita. Un vero e proprio ritardo mentale è presente solo nel 11- 17% dei casi con sindrome di Turner. Di frequente riscontro sono alcuni deficit conoscitivi, soprattutto il riconoscimento delle dimensioni dei rapporti dimensionali dagli oggetti. Sono frequenti disturbi della visione dei colori fino al daltonismo.
Diagnosi e terapia
La diagnosi di certezza si fonda sul reperto delle tipiche anomalie del corredo cromosomico.
Sono elementi di sospetto clinico:
- bassa statura (2,5 deviazioni standard sotto il valore medio per età)
- ritardo puberale in presenza di elevati livelli di gonadotropine plasmatiche e urinarie
Obiettivi della terapia della sindrome di Turner sono:
- la correzione dell’ infantilismo
- la correzione della bassa statura
- inserimento nella vita sociale.
La terapia sostitutiva con estrogeni deve essere a dosi sufficienti da rimuovere l’infantilismo e da promuovere la crescita, ma tale da non indurre precoce saldatura delle cartilagini epifisarie. L’ideale sarebbe mimare la normale secrezione estrogenica nel corso dello sviluppo. L’orientamento terapeutico attuale prevede la somministrazione di estrogeni (etinilestradiolo), utilizzando dosi di 0,1 mcg/die e in età prepuberale passante 5 mcg/die in età peripuberale, raddoppiando il dosaggio dopo i 13-14 anni con l’aggiunta ciclica di progesterone e passando successivamente a
una terapia con estroprogestinici. Tale terapia garantisce il mantenimento delle caratteristiche sessuali secondarie, la comparsa irregolare della mestruazione e la prevenzione dell’osteoporosi.
Ti potrebbero interessare:
- Sindrome di POEMS-Malattie rare
- Trisomia 18 o sindrome di Edwards
- Trisomia 13-Sindrome di Patau
- Malformazioni e malattie congenite: cause e prevenzione
Le informazioni contenute in questo sito non devono in alcun modo sostituire il rapporto medico-paziente;
si raccomanda di chiedere il parere del proprio medico prima di mettere in pratica qualsiasi consiglio o indicazione riportata.







