Policitemia vera
Definizione
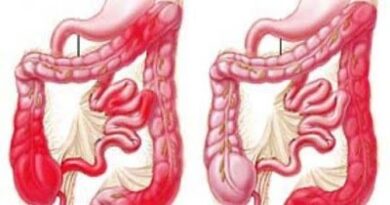
La policitemia vera (PV) è un disordine mieloproliferativo cronico caratterizzato da un’ alterazione clonale della cellula staminale con prevalente coinvolgimento dell’ eritropoiesi e conseguente espansione della massa eritrocitaria, un aumento dell’emoglobina (HB) e dell’ ematocrito (Hct). Tale espansione può interessare in minor misura anche le altre linee emopoietiche e dare origine a leucocitosi o trombocitosi. Letteralmente, policitemia significa “molte cellule nel sangue. Il nome policitemia significa appunto “più corpi cellulari sanguigni” rispetto al normale, ed è stato adottato per evidenziare la principale caratteristica di questa malattia, l’aumento nel sangue periferico degli eritrociti, dei globuli bianchi e delle piastrine.
Manifestazioni cliniche
La policitemia colpisce soggetti di età media di circa 60 anni, anche se sono descritti esordi in qualunque fascia di età. Predilige il sesso maschile con un rapporto M:F di 2:1. La diagnosi di policitemia può essere casuale, ma spesso è caratterizzata da sintomi minori dovuti all’aumento della viscosità ematica con prevalente interessamento del piccolo circolo quali:
In alcuni casi l’esordio si manifesta con processi trombociti o emorragici. Nel 15-20% dei casi l’esordio presenta una maggiore gravità clinica con fenomeni trombotici maggiori a livello di organi vitali. Si può infatti manifestare con:
- ictus cerebrale o TIA
- infarto miocardico acuto
- angina pectoris
- embolia polmonare
- infarti intestinali
Questi ultimi sono verosimilmente causati da micro-trombi del circolo gastroenterico e conseguente gastroduodenite erosiva o infarti mesenterici. Meno frequenti sono le trombosi della vena porta e delle vene sovraepatiche (sindrome di Budd-Chiari), che si presentano con:
- dolori addominali acuti
- dolore all’ipocondrio destro
- epatomegalia e ascite
Rare sono invece le manifestazioni emorragiche cutanee (porpora, ecchimosi, ematomi) e mucose (epistassi gengivorragia meno-metrorragie).
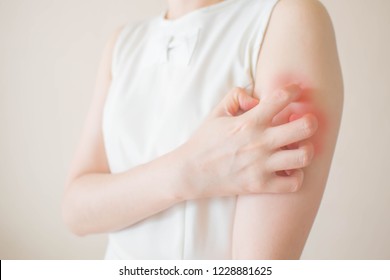
Un sintomo molto frequente è rappresentato ( 40% dei pazienti), dal prurito che si accenta soprattutto dopo il contatto con l’acqua calda e presenta la caratteristica peculiare di essere poco responsivo alla terapia con antistaminici. Frequenti sono anche le sudorazioni profuse, soprattutto notturne. All’esame obiettivo i pazienti affetti da policitemia presentano una colorazione rosso-vinoso al volto, naso, labbra e orecchie. La splenomegalia, causata dalla presenza di emopoiesi extramidollare o metaplasia mieloide, è presente in circa un terzo dei casi che generalmente è asintomatica all’esordio. Durante il decorso della malattia il graduale aumento dell’organo causa una sensazione di ripienezza gastrica e dolore all’ ipocondrio sinistro dovuti a infarti splenici.
Diagnosi
Il sospetto di policitemia deve nascere in primo luogo da un valore di Hct o di HB elevato. Se questo viene confermato in un successivo esame, il secondo passo è quello di eseguire la misura della massa eritrocitaria e del volume plasmatico. Una massa eritrocitaria aumentata e un volume plasmatico normale o aumentato sono compatibili con una diagnosi di policitemia, sia essa primaria o secondaria. Una massa eritrocitaria normale e un volume plasmatico diminuito sono invece caratteristici della policitemia relativa. Una volta documentato un incremento della massa eritrocitaria è necessario documentare la presenza degli altri criteri O.M.S per la diagnosi. Per una corretta diagnosi risultano utili la ricerca della clonalità basata sulla presenza di alterazioni genetiche, tra cui la presenza della mutazione del gene Jak2, la crescita di colonie spontanee dal midollo o dal sangue periferico e il livello di EPO sierica, generalmente basso nella forma primaria e alto in quelle secondarie. Contemporaneamente è necessario escludere, mediante esami ematochimici e strumentali la presenza di una policitemia acquisita secondaria a malattie neoplastiche o renali.
Criteri O.M.S. per la policitemia:
Criteri maggiori:
- HB >18,5 g/dl nell’uomo e >16,5 g/dl nella donna
- presenza della mutazione Jak2V617F o di mutazione dell’esone 12.
Criteri minori:
- mieloproliferazione delle tre linee cellulari all’esame istologico midollare
- livello di EPO sierica sotto il range di normalità
- crescita spontanea delle colonie eritroidi in vitro.
Per la diagnosi sono necessarie due criteri maggiori e un criterio minore oppure un criterio maggiore e due criteri minori.
Decorso e prognosi
La mortalità è legata soprattutto a episodi tromboembolici, più raramente emorragici, cui contribuiscono in certa misura l’età dei pazienti. In circa un terzo dei pazienti si osserva la trasformazione in un’altra malattia ematologica, principalmente in mielofibrosi (20%) e in leucemia mieloide acuta (15%). La sopravvivenza del paziente con policitemia è inficiata dalla comparsa di eventi trombotici e dalla trasformazione leucemica. Un buon controllo della massa eritrocitaria con periodiche flebotomie e un controllo dell’emopoiesi con una eventuale terapia citoriduttiva migliora notevolmente la prognosi permettendo lunghe sopravvivenze.
Terapia
Non appena posta la diagnosi la prima cosa da fare è cercare di diminuire al più presto il valore dell’ Hct per ridurre il rischio incombente di fenomeni tromboembolici fatali e debilitanti. Questo risultato si ottiene con flebotomia ripetuta , fino al raggiungimento di valori di Htc < 45%. Raggiunto questo valore è sufficiente mantenerlo con flebotomie periodiche. Nei casi più gravi attraverso una mielosoppressione praticata con Idrossiurea (HU) o alpha-Interferone (a-IFN). Quest’ultime terapie potrebbero però dare origine a effetti collaterali, anche gravi, e sono quindi riservate ai casi più difficili.
Ti potrebbero interessare: